Charakterystyka polimorfów leków metodą IGC- SEA
Wszystkie dane dla polimorfu A, w tym próbki mielone i mielone/nasycone, mieszczą się w granicach 49,9 mJ/m2. Natomiast polimorf B ma wyraźnie niższą dyspersyjną energię powierzchni wynoszącą 42,5 mJ/m2.
Wiadomo, że polimorf B jest termodynamicznie bardziej stabilną formą w warunkach otoczenia i chociaż inne czynniki również mają wpływ na ogólną stabilność, jest to zgodne z wynikami energii powierzchni. Jednak ani mielenie, ani obróbka wilgocią najwyraźniej nie powodują żadnej znaczącej zmiany energii powierzchniowej polimorfu A.
Uważa się, że mielenie polimorfu A wprowadza do materiału niewielką zawartość amorficzną, prawdopodobnie na powierzchni, obserwowaną w badaniach sorpcji wilgoci. Stwierdzono, że proces rekrystalizacji materiału z towarzyszącym temu procesowi wydalania wody następuje w zakresie od 50 do 60% RH. Aby zbadać to dokładniej, zmielony materiał badano w funkcji wilgotności względnej, zaczynając od 0% i stopniowo zwiększając RH. Próbce pozwolono na osiągnięcie równowagi przy każdej wilgotności przez około 6 godzin przed pomiarem czasu retencji.
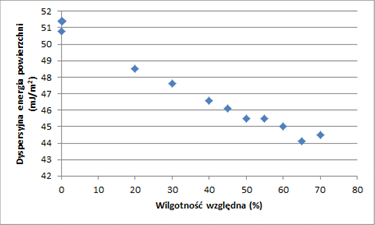
Wyniki tego badania przedstawiono na rysunkach 2 i 3. Rysunek 2 przedstawia składową dyspersyjną energii powierzchni zmielonej próbki jako funkcję wilgotności względnej w temperaturze 30°C. Oczywistym jest, że składowa niepolarna energii powierzchni stale maleje między 0 a 70% RH, gdy wzrasta ilość wody zaadsorbowanej na powierzchni. Jednak nie można zaobserwować żadnej nieciągłości między 50 a 60% RH, która mogłaby być powiązana z zapadnięciem się zawartości amorficznej. Ponadto po wystawieniu próbki na działanie 70% RH próbka została ponownie przetestowana przy 0% RH. Oba punkty przy 0% RH są przedstawione na rysunku 2 i są wyraźnie bardzo podobne. Nie można zaobserwować żadnej nieodwracalnej zmiany, co jest zgodne z energią powierzchniową zmielonej próbki wstępnie obrobionej przy 75% RH.
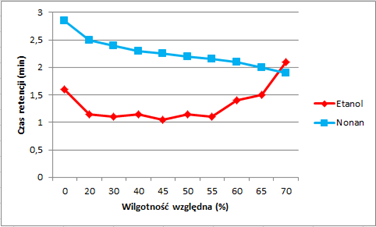
W przeciwieństwie do składowej dyspersyjnej energii powierzchni, interakcja zmielonej próbki z cząsteczkami polarnymi rozpuszczalników wykazuje wyraźną zmianę po wystawieniu na działanie wysokiej wilgotności. Rysunek 3 przedstawia czas retencji etanolu i nonanu (cząsteczka niepolarna o porównywalnym czasie retencji) wykreślony w zależności od wilgotności względnej. Podobnie jak w przypadku składowej niepolarnej energii powierzchni, nonan wykazuje stale malejący czas retencji, co jest zgodne ze zmniejszającą się interakcją przy wyższych wilgotnościach. Czas retencji etanolu również maleje do 50% RH, ale następnie wykazuje nagły i znaczący wzrost. Efekt ten zaobserwowano w innych układach, takich jak maltoza [1], w których obecność niewielkiej ilości materiału amorficznego powoduje wzrost czasu retencji z powodu dyfuzji masowej blisko temperatury zeszklenia (Tg). W tym przypadku eksperyment przeprowadzono w stałej temperaturze, ale pochłonięta wilgoć działa jak plastyfikator i powoduje spadek Tg. Wilgotność, przy której następuje zmiana, jest dokładnie tym samym punkcie, który zaobserwowano w danych sorpcji wody na aparacie DVS, w którym materiał rekrystalizuje w badaniach sorpcji pary. Powodem, dla którego efekt ten obserwuje się w przypadku etanolu, ale nie nonanu, jest to, że rozpuszczalnik polarny oddziałuje z próbką znacznie silniej niż rozpuszczalnik niepolarny. W skali czasowej eksperymentu iGC SEA nie obserwuje się żadnej krystalizacji materiału amorficznego. Można to postrzegać jako zmniejszenie czasu retencji.
Pokazuje to, że mielenie Polimorfu A rzeczywiście wprowadziło składnik amorficzny do materiału, ale nie zaobserwowano żadnej nieodwracalnej zmiany sugerującej zmianę morfologii w analizie próbki Polimorfu A, która została zmielona i rekrystalizowana przy 75% RH. W ramach czułości techniki nie można zaobserwować żadnych dowodów na to, że takie traktowanie powoduje rekrystalizację do Polimorfu B, pomimo wyższej stabilności termodynamicznej Polimorfu B.
Wnioski
iGC SEA okazał się czułym narzędziem do badania polimorfów farmaceutycznych. Energia powierzchni została wykorzystana do rozróżnienia i identyfikacji dwóch polimorfów tego samego leku oraz wykazano zgodność ze znanymi stabilnościami termodynamicznymi.
Pokazano, że zmielone próbki polimorfu A zawierają część materiału amorficznego a zeszklenie obserwowane w badaniach sorpcji następuje przy ok. 55% RH w 30°C. Obróbka wilgocią w celu rekrystalizacji tego materiału dała próbkę identyczną z oryginalnym polimorfem A, co sugeruje, że to podstawowa morfologia próbki, a nie stabilność termodynamiczna, napędza rekrystalizację.
Piśmiennictwo
1. Schultz, J., Lavielle, L. i Martin, C., J. Adhesion 23 (1987) 45


Bądź na bieżąco z nowościami z branży
Dołącz do odbiorców newslettera!